近日,我院刘春辉教授团队在国际知名期刊《The Journal of Physical Chemistry A》上发表题为“Competing Reaction Mechanisms and Origin of Selectivity in Base-Controlled (4 + 1) versus (2 + 1)/(4 + 2) Annulations of o-Aminochalcones with γ-Bromocrotonates”的重要理论研究成果。
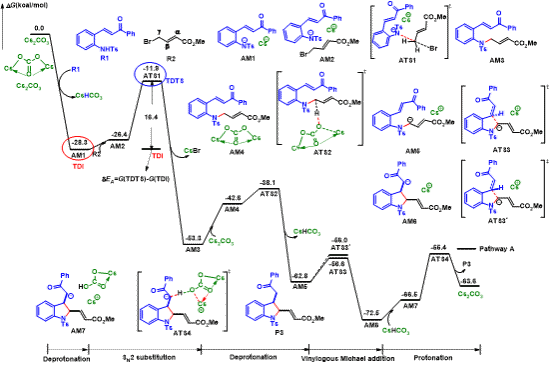
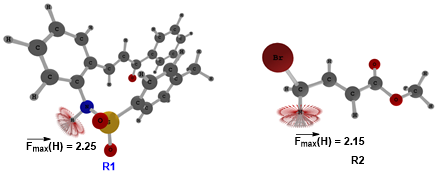
该研究通过高精度DFT计算、POCV反应性分析、NCI/AIM/NBO多维度工具系统揭示了Cs₂CO₃催化下γ-溴代巴豆酸酯与o-氨基查尔酮的(4+1)与(2+1)/(4+2)环化反应的竞争机制及选择性起源,明确解释了为何(4+1)环化路径是“胜利之路”,而另一条竞争路径则“此路不通”。研究发现,反应优先从o-氨基查尔酮的N–H键发生无势垒的去质子化,主要归因于该N–H键具有更强的酸性(pKa=19.3)和更高的原子反应性向量 (2.25),远优于γ-溴代巴豆酸酯中γ-C–H键的性能(pKa=32.8,原子反应性向量
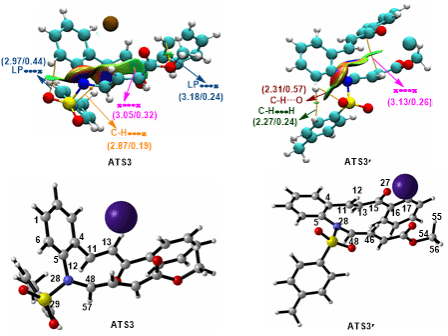
(2.25),远优于γ-溴代巴豆酸酯中γ-C–H键的性能(pKa=32.8,原子反应性向量 2.15)。能量跨度分析进一步显示,(4+1)环化路径的能隙仅为16.4 kcal/mol,而(2+1)/(4+2)环化路径中的整体能隙则高达30.2 kcal/mol。并且在后者路径中,一旦通过γ-C–H去质子化启动,后续分子内SN2取代的能垒将陡升至21.7 kcal/mol,且关键中间体BM8比其前体BM7高出13.8 kcal/mol,极易发生逆反应,因此该路径在动力学与热力学上均明显不利。在非对映选择性方面,顺式过渡态ATS3因存在两个LP···π、一个C–H···π和一个π···π相互作用构成的协同稳定非共价网络,以及LP(C48)→BD*(C11–C13)轨道相互作用带来的高达79.82 kcal/mol二级微扰能,使其比反式过渡态ATS3′能量低0.6 kcal/mol,从而成功解释了实验中43% de的选择性。该工作不仅从分子层面揭示了反应化学选择性与非对映选择性的本质,也展示了理论化学为实验“画像”、为设计“导航”的示范价值,为未来碱催化环化反应的理性设计提供了精确的分子蓝图。
2.15)。能量跨度分析进一步显示,(4+1)环化路径的能隙仅为16.4 kcal/mol,而(2+1)/(4+2)环化路径中的整体能隙则高达30.2 kcal/mol。并且在后者路径中,一旦通过γ-C–H去质子化启动,后续分子内SN2取代的能垒将陡升至21.7 kcal/mol,且关键中间体BM8比其前体BM7高出13.8 kcal/mol,极易发生逆反应,因此该路径在动力学与热力学上均明显不利。在非对映选择性方面,顺式过渡态ATS3因存在两个LP···π、一个C–H···π和一个π···π相互作用构成的协同稳定非共价网络,以及LP(C48)→BD*(C11–C13)轨道相互作用带来的高达79.82 kcal/mol二级微扰能,使其比反式过渡态ATS3′能量低0.6 kcal/mol,从而成功解释了实验中43% de的选择性。该工作不仅从分子层面揭示了反应化学选择性与非对映选择性的本质,也展示了理论化学为实验“画像”、为设计“导航”的示范价值,为未来碱催化环化反应的理性设计提供了精确的分子蓝图。
本工作许昌学院为第一单位,韩培林博士为第一作者,2022级应用化学专业本科生江宇博、闫永利为第二、第三作者,刘春辉教授为通讯作者,并得到了国家自然科学基金的支持。
论文链接:https://doi.org/10.1021/acs.jpca.5c08373 (一审:祁宜宜 二审:李航 三审:何伟伟)




